
Diagnóstico Bioquímico y Molecular de Enfermedades Hereditarias
Somos expertos en el abordaje integral del paciente para el diagnóstico de enfermedades hereditarias.
Las enfermedades metabólicas hereditarias (EMH) se engloban dentro de las enfermedades raras o poco prevalentes. Las EMH son enfermedades genéticas debidas a mutaciones en un gen que codifica para una proteína que interviene en una vía metabólica. La función anormal de la proteína implicada puede causar acúmulo de sustancias y/o deficiencia de otras que son necesarias para el organismo, causando así enfermedad. Los síntomas pueden ser heterogéneos, crónicos, multisistémicos y progresivos, pudiéndose presentar a cualquier edad de la vida. En la actualidad, se han descrito alrededor de unas mil entidades, pero exiten pacientes que no disponen de diagnóstico definitivo y/o tratamiento eficaz.
Es un área emergente en la cual la investigación y las nuevas tecnologías llevan a la descripción de nuevas enfermedades de forma constante, por lo que el diagnóstico y la búsqueda de nuevas terapias esta siendo un reto importante en salud a nivel mundial.
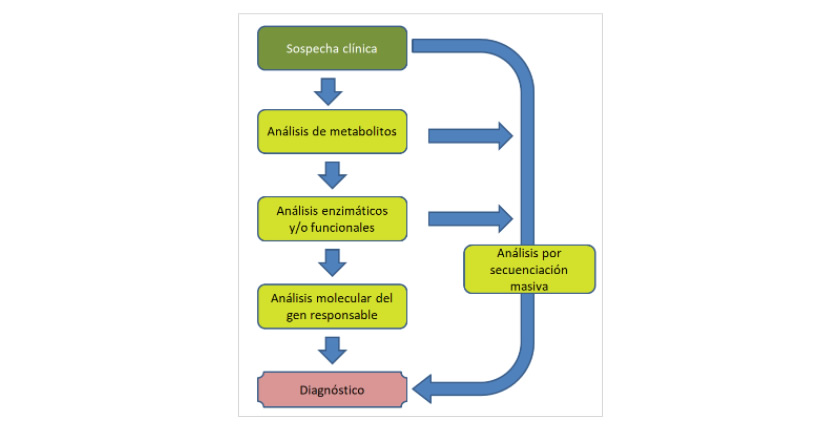
Ante una sospecha clínica de que un paciente pueda ser afecto de una EMH, el proceso a seguir para llegar a un diagnóstico es:
Análisis de metabolitos
Las diferentes tecnologías de cromatografía y espectrometría de masas nos han permitido desarrollar diferentes metodologías para la valoración de biomarcadores específicos para el diagnóstico y monitorización de tratamiento de las EMH. Se realizan, entre otros, el estudio de los siguientes metabolitos:
- Ácidos orgánicos en orina
- Lisoesfingolípidos lisoGb1, lisoGb3, LisoSM, lisoSM-509
- Lisofosfatidilcolinas C22, C24 y C26
- Cistina intraleucocitaria
- Acilcarnitinas
- Aminoácidos
- Oxiesteroles
- Ácidos grasos de cadena muy larga
- Esteroles
- Isoformas de las sialotransferrinas
- Tipificación de glucosaminoglicanos
- Oligosacáridos
La alteración de estos metabolitos orientan al diagnóstico de una patología o grupo de patologías para poder proseguir con los siguientes análisis :
Análisis enzimáticos y/o funcionales
Realizamos actividades enzimáticas para identificar defectos concretos o para orientar el diagnóstico, como por ejemplo:
- Actividades de enzimas lisosomales
- Análisis de las actividades de los complejos de la cadena respiratoria mitocondrial
- Estudio de la Piruvato deshidrogenasa
- Análisis de la oxidación de sustratos
- Oxidación de palmitato deuterado
- Estudio de galactosemias
Si se detecta una patología que sólo puede ser debida a la deficiencia de un gen concreto, recurrimos al:
Análisis molecular por secuenciación Sanger
Tenemos disponible en nuestro catálogo la secuenciación por método Sanger de unos 40 genes relacionados con EMH, para estudio de casos índice, familiares con mutación conocida y para el diagnóstico prenatal en los casos donde se conoce/n la/s mutación/nes familiar/es.
Análisis molecular por secuenciación masiva
Abordamos el estudio de una gran variedad de enfermedades hereditarias mediante la realización del exoma celular completo. Hemos agrupado los genes relacionados con EMH en paneles virtuales según la vía metabólica afectada, las características clínicas o los metabolitos alterados, lo que permite un análisis dirigido al grupo de genes candidatos más rápido y con más eficiencia. En este sentido, actualmente, tenemos una eficacia de resolución del 65% en presencia de biomarcadores positivos. Se trata de una tecnología de secuenciación de alto rendimiento. Algunos ejemplos de los paneles son:
.
- Genes peroxisomales
- Genes lisosomales
- Genes acidemias metilmalónicas
- Genes beta-oxidación mitocondrial
- Genes de aminoácidopatías y acidurias orgánicas
- Genes fenilcetonuria e hiperfenilalaninemias
- Genes glicosilación de proteinas: CDG
- Genes ciclo de la urea y relacionados
La secuenciación masiva se puede utilizar en todos los casos en los que, dependiendo de la clínica del paciente, los marcadores bioquímicos y/o las actividades enzimáticas, haya más de un gen que pueda ser el causante de la patología del paciente.
En algunos casos en los que se identifiquen variantes de significado incierto (VUS) en algún gen candidato, y no se haya hecho la caracterización bioquímica, puede ser necesario recurrir al estudio de metabolitos, y/o actividades enzimáticas para confirmar el posible efecto de estas variantes. Además, es importante destacar, que en el caso de no identificar el defecto genético, o ante un nuevo hallazgo bioquímico o clínico, es posible reanalizar los datos con los paneles virtuales que se crea necesarios, dando más facilidad para llegar al diagnóstico de la patología del paciente.
Diagnóstico prenatal de EMH
En caso de tener antecedentes familiares de una EMH concreta, nuestro centro ofrece el diagnóstico prenatal de nuevas gestaciones de la familia. Para ello utilizamos el análisis enzimático y/o el estudio molecular de las mutaciones familiares en el gen afectado, escogiendo la estrategia más adecuada según la enfermedad, la información previa del caso índice, edad gestacional y antecedentes.
Otras enfermedades metabólicas
Análisis bioquímico y molecular de la Porfirias
Las porfirias son un grupo heterogéneo de enfermedades metabólicas causadas por alteraciones en la biosíntesis del grupo hemo. Cada tipo de porfiria se caracteriza por un defecto en una enzima diferente involucrada en esta vía, ocasionando un patrón específico de acumulación de porfirinas y sus precursores. Los síntomas que presentan los pacientes son muy variados entre las distintas enfermedades, causando mayoritariamente afectaciones en la piel, el hígado y el sistema eritropoyético. Algunas porfirias también pueden manifestarse en forma de ataques neurológicos agudos potencialmente letales.
Las deficiencias enzimáticas son debidas a mutaciones en los genes que codifican para las enzimas correspondientes, por lo que el diagnóstico de las porfirias debe abordarse desde el punto de vista bioquímico y genético, y debe realizarse de manera secuencial.
Además, nuestro laboratorio forma parte de la EPNET (European Porphyria Network).
Cada tipo de porfiria se expresa con un patrón metabólico en orina, sangre y heces característico que puede ser tipificado por fluorimetria y cromatografía líquida. Los patrones de porfirinas obtenidos en diferentes líquidos biológicos permiten el diagnóstico y la clasificación de las porfirias con elevada precisión. El diagnóstico de ataques agudos se establece midiendo ácido delta aminolevulinico (ALA) urinario y porfobilinógeno (PBG) en orina. Una vez tipificada bioquímicamente el tipo de porfiria se puede realizar el estudio genético en búsqueda de la mutación causante.
Una vez identificada la enzima disfuncional se realiza el estudio molecular completo mediante secuenciación directa y si fuera necesario por MLPA.
Análisis integrado de la enfermedad de Wilson
La enfermedad de Wilson es una enfermedad congénita caracterizada por la acumulación tóxica en el organismo de cobre procedente de la dieta, especialmente en el hígado, en el cerebro y el ojo, en este último formando los anillos de Kayser-Fleischer. La enfermedad de Wilson se manifiesta principalmente como enfermedad hepática y/o neurológica. El diagnóstico precoz de esta enfermedad es fundamental, debido a la elevada morbi-mortalidad asociada a esta entidad y a la existencia de un tratamiento específico muy eficaz.
La enfermedad de Wilson es una enfermedad hereditaria autosómica recesiva, causada por la alteración de ambas copias del gen ATP7B. Este gen codifica una proteína necesaria para eliminar el cobre sobrante desde el interior de la célula hepática a la bilis.
Estudio bioquímico:
El diagnóstico bioquímico se basa en la detección de concentraciones anormalmente bajas de cobre en suero y elevadas en orina de 24 horas por espectrometría de absorción atómica. La ceruloplasmina, proteína transportadora de cobre, también es un biomarcador útil, y en la enfermedad de Wilson se encuentra en concentraciones plasmáticas bajas. Alternativamente, se pueden detectar concentraciones elevadas de cobre en biopsia hepática.
Estudio molecular:
Se realiza en análisis molecular completo del gen ATP7B mediante secuenciación directa y MLPA.