
Diagnóstico Bioquímico y Molecular de Enfermedades Hereditarias
Somos expertos en el abordaje integral del paciente para el diagnóstico de enfermedades hereditarias.
Biopsia líquida: estudio de ADN tumoral libre circulante
El Hospital Clínic es centro pionero en la incorporación de herramientas moleculares no invasivas para el seguimiento de pacientes con cáncer de pulmón. El estudio en ADN tumoral libre circulante de mutaciones específicas en el gen del receptor del factor de crecimiento epidérmico (EGFR) en muestra de plasma, permite estratificar al paciente y monitorizar la respuesta al tratamiento en pacientes con cáncer de pulmón de células no pequeñas. Esta técnica beneficia a aquellos pacientes donde no es posible obtener una biopsia o en pacientes con múltiples metástasis donde biopsiar cada una de ellas sería impracticable.
Espectrometría de masas en tándem
La cromatografía líquida acoplada a la espectrometría de masas en tándem (LC-MS/MS) es una potente técnica analítica que ha revolucionado el laboratorio clínico en estos últimos años. Presenta grandes ventajas permitiendo analizar con muy poca cantidad de muestra un amplio espectro de metabolitos en un tiempo corto, que a su vez se pueden identificar y cuantificar con una gran selectividad, sensibilidad y especificidad.

La aplicación de esta tecnología nos ha permitido ampliar el cribado neonatal de Cataluña, de 3 a 20 enfermedades, así como cuantificar diversos biomarcadores para el diagnóstico y monitorización de tratamiento de más de 400 entidades genéticas. Con esta metodología también hemos podido sustituir técnicas radiométricas obteniendo la misma sensibilidad y una mejor especificidad, así como eliminar técnicas obsoletas que requerían una gran cantidad de volumen de muestra.
Secuenciación masiva del exoma completo
La secuenciación masiva del exoma completo permite obtener las variantes de todas las regiones génicas que codifican para proteínas. Aunque el exoma representa menos del 2% de todo el genoma (~45Mb), se estima que contiene el 85% de las variantes causantes de enfermedades mendelianas. Se trata de una tecnología de secuenciación de alto rendimiento.
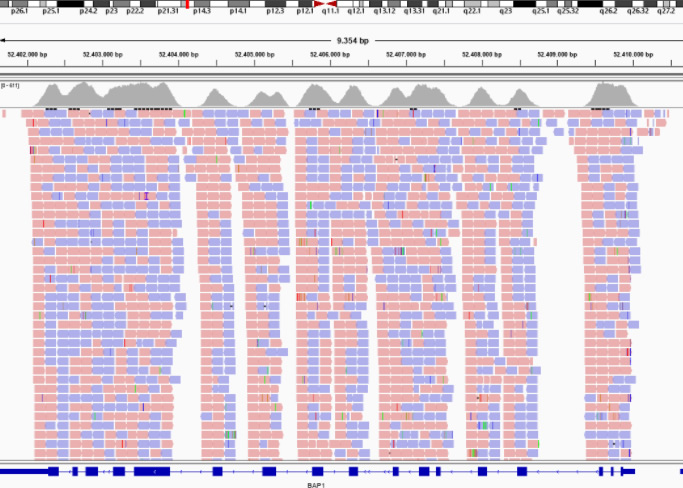
En nuestro hospital utilizamos el protocolo de secuenciación masiva Nextera flex for Enrichment con sondas Exoma completo (Illumina) y posterior secuenciación en la plataforma NextSeq550 (Illumina). Disponemos de un servicio de análisis bioinformático de los datos. Posteriormente un facultativo realiza el análisis genético. En base a la información clínica facilitada se filtra por paneles de genes definidos (por ejemplo Hipertensión pulmonar, cardiomiopatías familiares o enfermedades metabólicas) o se crea un panel virtual específico (por ejemplo pacientes con discapacidad intelectual que presentan dismorfia u otros fenotipos asociados, o pacientes con enfermedades muy raras con una prevalencia < 1/1.000.000). Finalmente, se emite un informe con los resultados hallados, su interpretación y asesoramiento genético.
Los datos generados se almacenan y se ofrece la oportunidad de realizar ampliación de estudio en los casos que lo requieran (por ejemplo: nuevos genes identificados asociados a la enfermedad o aparición de otros fenotipos con origen genético). Una vez detectada la causa genética, se puede proceder al estudio de los familiares a riesgo y proporcionar un adecuado asesoramiento genético.
Secuenciación masiva por paneles de genes
El fundamento de la secuenciación masiva de paneles de genes es el mismo que en el caso del exoma. El uso de secuenciación masiva de paneles de genes permite reducir costes en comparación con la secuenciación del exoma completo. Este estudio permite (según el diseño) incorporar otras regiones de interés más allá de los exones y regiones intrónicas flanqueantes, presenta coberturas mayores de las regiones analizadas, pero ofrece un número reducido de genes a analizar, que puede variar dependiendo de su tamaño y el diseño de cada panel.
En la actualidad ofrecemos el estudio del panel de cáncer, el panel de hiperlipidemias o el panel MODYs entre otros. En todos los casos, un facultativo realiza el análisis genético y emite un informe con los resultados hallados, su interpretación y asesoramiento genético.
Secuenciación Sanger

Se trata de un método de secuenciación que consiste en amplificar un fragmento de interés mediante PCR y posteriormente realizar una nueva reacción en la que se incorporan selectivamente nucleótidos terminadores marcados con fluorescencia mezclados con nucleótidos normales. Esto da lugar a fragmentos de distintos tamaños que terminan con un nucleótido marcado con fluorescencia. El producto de esta reacción se somete a una electroforesis capilar con lectura simultánea de fluorescencia, en la que los fragmentos se ordenan por tamaño y se obtiene un electroferograma de fluorescencia que se transforma en una secuencia de nucleótidos mediante un software de análisis específico. Este proceso permite el análisis de un fragmento pequeño de ADN (300-1000b) y la detección de variantes puntuales y pequeñas inserciones/deleciones con alta fiabilidad.
MLPA
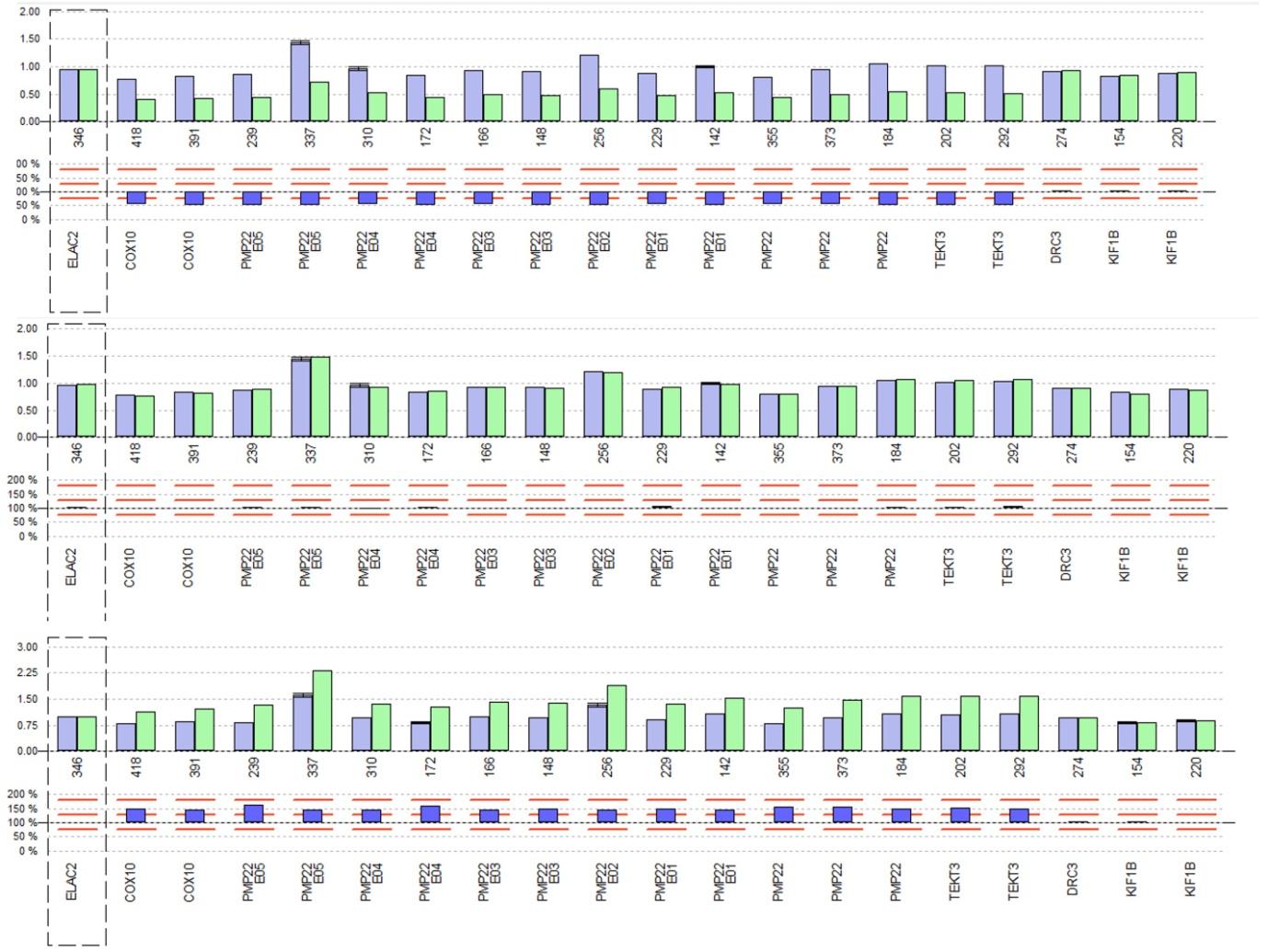
Se trata de una variante de la PCR, en la que se amplifican múltiples secuencias diana a las que se añaden cebadores específicos (sondas) que se unen de forma específica a la secuencia de interés y tienen un tamaño distinto conocido marcadas con fluorescencia. Estos fragmentos de distintos tamaños se amplifican y posteriormente se someten a una electroforesis capilar con lectura simultánea de fluorescencia. En este caso se obtiene un perfil que muestra picos de distinto tamaño con distinta área bajo la curva. Comparando muestras de pacientes a estudio con muestras control podemos identificar: duplicaciones (mayor área bajo la curva en pacientes que en controles), deleciones (menor área bajo la curva en pacientes que en controles) o identificar presencia/ausencia de variantes concretas. En una reacción de MLPA podemos analizar hasta ~50 fragmentos diferentes por reacción incluyendo análisis de ganancias o pérdidas de material genético y/o análisis de variantes específicas.
Hibridación genómica comparativa o CGH
La hibridación genómica comparativa o CGH (por sus siglas en inglés Comparative Genomic Hybridization) es un método de citogenética molecular empleado para analizar variaciones del número de copias (copy number variants o CNV). Relaciona el nivel de ploidía del ADN de una muestra en comparación con una muestra de referencia sin la necesidad de realizar un cultivo celular. El objetivo de esta técnica es comparar de manera rápida y eficiente dos muestras de ADN genómico para detectar ganancias o pérdidas de material genético.
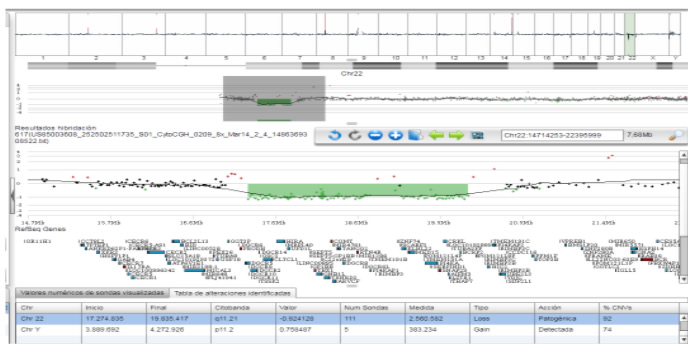
El aCGH empleado en nuestro laboratorio se compone de más de 60.000 sondas de tipo oligonucleótido. Este diseño permite interrogar a más de 300 regiones del genoma causantes de reordenamientos recurrentes relacionados con síndromes genómicos conocidos con una alta cobertura (en promedio 1 sonda/10Kb), ofreciendo un tiempo de respuesta en muestras prenatales de 10-15 días laborables desde la recepción de la muestra.
Estudio de expansión de tripletes (TP-PCR)
La TP-PCR es una modificación de la PCR, específica para el estudio de zonas repetidas. En ella se utiliza un cebador adyacente a la zona repetida y otro cebador diseñado sobre la repetición que tiene añadida una cola que no hibrida con el ADN molde. Esta cola es complementaria a un tercer cebador denominado universal. Los productos obtenidos mediante la TP-PCR se analizan mediante electroforesis capilar.
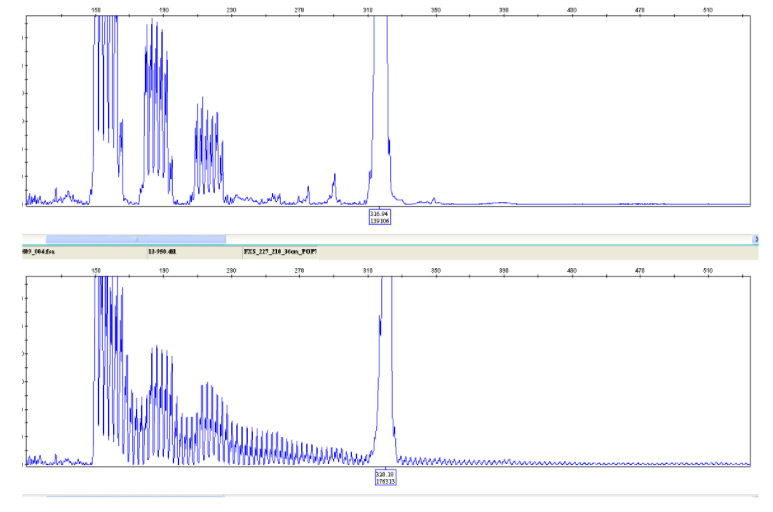
En el caso de que exista expansión, se obtiene una imagen electroforética típica de escalera, en la que cada pico corresponde a una de las repeticiones de la expansión. Esta técnica permite detectar grandes ampliaciones incluso cuando no se pueden amplificar por PCR convencional, lo que mejora el diagnóstico de este tipo de enfermedades.
Realización de Estudios funcionales para la caracterización de variantes de significado incierto (VUS) en enfermedades metabólicas hereditarias
Hemos desarrollado diferentes estudios funcionales en fibroblastos (obtenidos de biopsia de piel u otros tejidos) para demostrar el bloqueo de una determinada vía metabólica, concretamente para el diagnóstico de defectos del metabolismo del piruvato, de enfermedades mitocondriales y de deficiencias de la beta-oxidación mitocondrial. Además nos permite confirmar el efecto de mutaciones de significado incierto encontradas para dichas entidades.