
Diagnòstic Bioquímic i Molecular de Malalties Hereditàries
Som experts en abordatge integral del pacient per al diagnòstic de malalties hereditàries.
Les malalties metabòliques hereditàries (EMH) s'engloben dins de les malalties rares o poc prevalents. Les EMH són malalties genètiques degudes a mutacions en un gen que codifica per a una proteïna que intervé en una via metabòlica. La funció anormal de la proteïna implicada pot causar acumulació de substàncies i/o deficiència d'altres que calen per a l'organisme, causant així malaltia. Els símptomes poden ser heterogenis, crònics, multisistèmics i progressius, podent presentar-se a qualsevol edat de la vida. Actualment, s'han descrit al voltant d'unes mil entitats, però hi ha pacients que no disposen de diagnòstic definitiu i/o tractament eficaç.
És una àrea emergent on la recerca i les noves tecnologies porten a la descripció de noves malalties de forma constant, per la qual cosa el diagnòstic i la recerca de noves teràpies està sent un repte important en salut a nivell mundial.
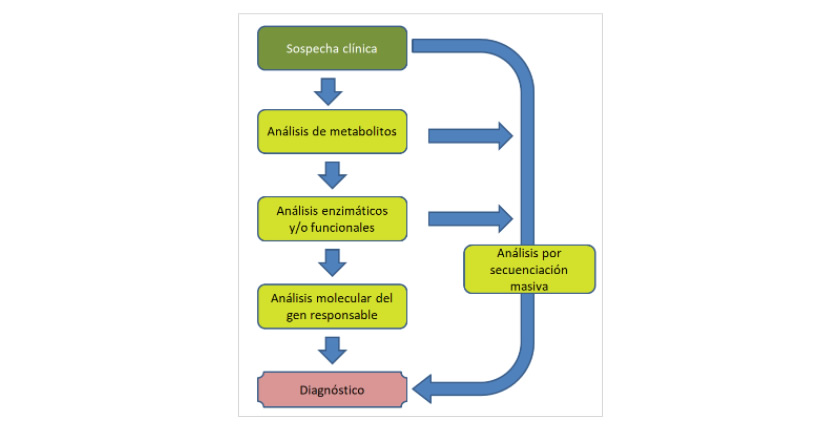
Davant d'una sospita clínica que un pacient pugui ser afecte d'una EMH, el procés que cal seguir per arribar a un diagnòstic és:
Anàlisi de metabòlits
Les diferents tecnologies de cromatografia i espectrometria de masses ens han permès desenvolupar diferents metodologies per a la valoració de biomarcadors específics per al diagnòstic i monitorització de tractament de les EMH. Es realitzen, entre d'altres, l'estudi dels metabòlits següents:
- Àcids orgànics en orina
- Lisoesfingolípids lisoGb1, lisoGb3, LisoSM, lisoSM-509
- Lisofosfatidilcolines C22, C24 i C26
- Cistina intraleucocitària
- Acilcarnitines
- Aminoàcids
- Oxiesterols
- Àcids grassos de cadena molt llarga
- Esterols
- Isoformes de les sialotransferrines
- Tipificació de glucosaminoglicans
- Oligosacàrids
L'alteració d'aquests metabòlits orienten el diagnòstic d'una patologia o grup de patologies per poder continuar amb les anàlisis següents:
Anàlisis enzimàtiques i/o funcionals
Fem activitats enzimàtiques per identificar defectes concrets o per orientar el diagnòstic, com ara:
- Activitats d'enzims lisosomals
- Anàlisi de les activitats dels complexos de la cadena respiratòria mitocondrial
- Estudi de la Piruvat deshidrogenasa
- Anàlisi de l'oxidació de substrats
- Oxidació de palmitat deuterat
- Estudi de galactosèmies
Si es detecta una patologia que només pot ser deguda a la deficiència dun gen concret, recorrem al:
Anàlisi molecular per seqüenciació Sanger
Tenim disponible al nostre catàleg la seqüenciació per mètode Sanger d'uns 40 gens relacionats amb EMH, per a estudi de casos índex, familiars amb mutació coneguda i per al diagnòstic prenatal en els casos on es coneix/en la/les mutació/ns familiar/s.
Anàlisi molecular per seqüenciació massiva
Abordem l'estudi d'una gran varietat de malalties hereditàries mitjançant la realització del exoma cel·lular complet. Hem agrupat els gens relacionats amb EMH en panells virtuals segons la via metabòlica afectada, les característiques clíniques o els metabòlits alterats, fet que permet una anàlisi dirigida al grup de gens candidats més ràpid i amb més eficiència. En aquest sentit, actualment tenim una eficàcia de resolució del 65% en presència de biomarcadors positius. És una tecnologia de seqüenciació d'alt rendiment. Alguns exemples dels panells són:
.
- Gens peroxisomals
- Gens lisosomals
- Gens acidèmies metilmalòniques
- Gens beta-oxidació mitocondrial
- Gens d'aminoàcidpaties i aciduries orgàniques
- Gens fenilcetonúria i hiperfenilalaninèmies
- Gens glicosilació de proteïnes: CDG
- Gèns cicle de la urea i relacionats
La seqüenciació massiva es pot utilitzar en tots els casos en què, depenent de la clínica del pacient, els marcadors bioquímics i/o les activitats enzimàtiques, hi hagi més d'un gen que pugui ser el causant de la patologia del pacient.
En alguns casos en què s'identifiquin variants de significat incert (VUS) en algun gen candidat, i no s'hagi fet la caracterització bioquímica, pot ser necessari recórrer a l'estudi de metabòlits i/o activitats enzimàtiques per confirmar el possible efecte d'aquestes variants. A més, és important destacar, que en el cas de no identificar el defecte genètic, o davant d'una nova troballa bioquímica o clínica, és possible reanalitzar les dades amb els panells virtuals que es cregui necessaris, donant més facilitat per arribar al diagnòstic de la patologia del pacient.
Diagnòstic prenatal d'EMH
En cas de tenir antecedents familiars duna EMH concreta, el nostre centre ofereix el diagnòstic prenatal de noves gestacions de la família. Per fer-ho utilitzem l'anàlisi enzimàtica i/o l'estudi molecular de les mutacions familiars al gen afectat, escollint l'estratègia més adequada segons la malaltia, la informació prèvia del cas índex, edat gestacional i antecedents.
Altres malalties metabòliques
Anàlisi bioquímica i molecular de la Porfíries
Les porfíries són un grup heterogeni de malalties metabòliques causades per alteracions a la biosíntesi del grup hemo. Cada tipus de porfíria es caracteritza per un defecte en un enzim diferent involucrat en aquesta via, ocasionant un patró específic d'acumulació de porfirines i els seus precursors. Els símptomes que presenten els pacients són molt variats entre les diferents malalties, causant majoritàriament afectacions a la pell, el fetge i el sistema eritropoiètic. Algunes porfíries també es poden manifestar en forma d'atacs neurològics aguts potencialment letals.
Les deficiències enzimàtiques són degudes a mutacions en els gens que codifiquen per als enzims corresponents, per la qual cosa el diagnòstic de les porfíries s'ha d'abordar des del punt de vista bioquímic i genètic, i s'ha de fer de manera seqüencial.
A més, el nostre laboratori forma part de l'EPNET (European Porphyria Network).
Estudi bioquímic:
Cada tipus de porfíria s'expressa amb un patró metabòlic en orina, sang i femta característica que pot ser tipificada per fluorimetria i cromatografia líquida. Els patrons de porfirines obtinguts en diferents líquids biològics permeten el diagnòstic i la classificació de les porfíries amb elevada precisió. El diagnòstic d'atacs aguts s'estableix mesurant àcid delta aminolevulinic (ALA) urinari i porfobilinogen (PBG) en orina. Un cop tipificada bioquímicament el tipus de porfíria es pot fer l'estudi genètic a la recerca de la mutació causant.
Estudi molecular:
Un cop identificat l'enzim disfuncional es realitza l'estudi molecular complet mitjançant seqüenciació directa i, si cal, per MLPA.
Anàlisi integrada de la malaltia de Wilson
La malaltia de Wilson és una malaltia congènita caracteritzada per la acumulació tòxica a l'organisme de coure procedent de la dieta, especialment al fetge, al cervell i l'ull, en aquest últim formant els anells de Kayser-Fleischer. La malaltia de Wilson es manifesta principalment com a malaltia hepàtica i/o neurològica. El diagnòstic precoç d'aquesta malaltia és fonamental, degut a l'elevada morbimortalitat associada a aquesta entitat ia l'existència d'un tractament específic molt eficaç.
La malaltia de Wilson és una malaltia hereditària autosòmica recessiva, causada per l'alteració de les dues còpies del gen ATP7B. Aquest gen codifica una proteïna necessària per eliminar el coure sobrant des de l'interior de la cèl·lula hepàtica a la bilis.
Estudi bioquímic:
El diagnòstic bioquímic es basa en la detecció de concentracions anormalment baixes de coure en sèrum i elevades en orina de 24 hores per espectrometria d'absorció atòmica. La ceruloplasmina, proteïna transportadora de coure, també és un biomarcador útil, i en la malaltia de Wilson es troba a concentracions plasmàtiques baixes. Alternativament, es poden detectar concentracions elevades de coure en biòpsia hepàtica.
Estudi molecular:
Es realitza en anàlisi molecular completa del gen ATP7B mitjançant seqüenciació directa i MLPA.
Proves de catàleg
Anàlisi de metabòlits a EMH
Disposem de més de 60 proves diferents per a anàlisis de biomarcadors, alguns exemples són:
- Àcids orgànics en orina – Codi 16075
- Acilcarnitines– Codi 16301
- Lisoesfingolípids lisoGb1, lisoGb3, LisoSM, lisoSM-509 – Codi 16387
- Lisofosfatidilcolines C22, C24 i C26 – Codi 16384
- Aminoàcids– Codi 16078
- Oxiesterols en plasma– Codi 16341
- Àcids grassos de cadena molt llarga en sèrum– Codi 16059
- Esterols– Codi 16131
- Isoformes de sialotransferrines-Codi 16134
- Tipificació de glucosaminoglicans – Codi 16016
- Oligosacàrids– Codi 16015
- Galactosa (Galactosèmies), plasma – Codi 16200
- Cistina intraleucocitària – Codi 16088
Anàlisis enzimàtiques i/o funcionals a EMH
Disposem de més de 80 proves d'activitats d'enzims lisosomals en diferents materials com sèrum, sang seca, leucòcits o fibroblasts. Algunes són:
- Alfa-Galactosidasa A (E. Fabry) – Codi 16187, 16143, 16006
- Beta-Galactosidasa (Gangliosidosi GM1 i Morquio B) - Codi 16173, 16189
- Beta-Glucocerebrosidasa (E. Gaucher) – Codi 16337, 16004,
- Arilsulfatasa A (Leucodistròfia metacromàtica) – Codi 16007, 16150
- Palmitoil proteïna tioesterasa (Ceroide Lipofuscinosi Neuronal Infantil_CNL1)-Cód. 16000, 16172
- Heparan-N-sulfatasa (E. Sanfilippo A) – Codi 16023, 16162
- Iduronosulfatasa (E. Hunter) – Codi 16164, 16165
A més, disposem d'altres proves relacionades amb malalties mitocondrials i altres EMH:
- Anàlisi d'activitat dels complexos de la cad. resp. mitocondrial, fibroblasts – Codi 16154
- Anàlisi de les activitats dels complexos de la cad. resp. mitocondrial, múscul – Codi 16102
- Estudi de la Piruvat deshidrogenasa, fibroblasts– Codi 16272
- Anàlisi de l'oxidació de substrats, fibroblasts– Codi 16273
- Oxidació de palmitat deuterat, fibroblasts– Codi 16094
Anàlisi molecular per seqüenciació Sanger a EMH
Oferim l'anàlisi molecular de més de 40 gens relacionats amb EMH. Alguns exemples:
- Gen ABCD1 (Adrenoleucodistròfia lligada al X) - recerca de mutacions, sang – Cód. 16140
- Gen CYP27A1 (Xantomatosi cerebrotendinosa)-cerca. de muts (cas índex), sang–Cód. 16260
- Gen GAA (E. Pompe)- cerca de mutacions (cas índex), sang total- Codi 16184
- Gen GLA (E. Fabry) - recerca de mutacions, sang total – Codi 16134
- Gen IDUA (E. Hurler)- cerca de mutacions, sang total – Codi 16183
Anàlisi molecular per seqüenciació massiva
Anàlisi de l'exoma mitjançant panells virtuals de gens. Disposem de més de 40 panells que cobreixen més de 700 gens diferents relacionats amb EMH. Alguns exemples:
- Gens peroxisomals – Codi 16350, 16349
- Gens lisosomals – Codi 16363, 16362, 16361
- Gens acidèmies metilmalòniques– Codi 16378
- Gens altres aminoàcidpaties i aciduries orgàniques – Codi 16379
- Gens beta-oxidació mitocondrial– Codi 16375, 16374
- Gens fenilcetonúria i hiperfenilalaninèmies – Codi 16370
- Gens glicosilació de proteïnes: CDG – Codi 16368, 16369
- Gèns cicle de la urea i relacionats – Codi 16371
Porfíries
- Estudi de porfirines en sang, orina, femta – Codis 80142, 80140, 80141
- Porfíria- cerca de mutacions (cas índex), sang total - Codi 70223
- Porfiria- mutació concreta (cas familiar), sang total – Codi 71000
Malaltia de Wilson
- Cobre en sèrum, orina 24h, teixit – Codis 50157, 50558, 5861
- Malaltia de Wilson- cerca de mutacions (cas índex i portador no relacionat amb la família), sang total Codi 70200
- Malaltia de Wilson- mutació concreta (cas familiar), sang total – Codi 70222
Actualitat

Durant aquesta temporada, l'Hospital Clínic de Barcelona ha

El món del llibre s'uneix per recaptar fons per a la investigació en immunoteràpia de càncer d'Hospital Clínic-IDIBAPS